庞贝氏病(糖原贮积症II型)
时间:2020年07月02日信息来源:本站原创点击:次
糖原贮积症Ⅱ型(GSDⅡ)也称α-葡萄糖苷酶缺乏症,由1932年荷兰病理学家Pompe首次报道,故常称为庞贝氏病(Pompe病),是一种罕见的常染色体隐性遗传的进展性溶酶体病,也是目前所知唯一属于溶酶体病的糖原贮积症。发病率约为1/40000~1/50000活产婴儿,但存在种族及地区差异。
GSD II临床表现
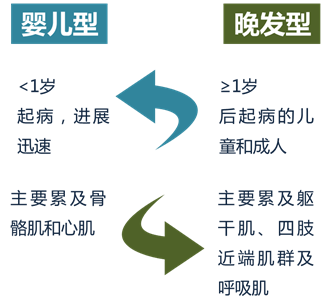
是累及全身的系统性疾病,临床变异较大,但主要以肌病表现为主。根据发病年龄、受累器官和疾病进展速度,临床上将GSDⅡ分为婴儿型和晚发型两大类:
GSD II发病机制
GAA基因突变导致溶酶体内的α-葡萄糖苷酶活性缺乏或显著降低,糖原不能被降解而沉积在骨骼肌、心肌和平滑肌等细胞的溶酶体内,导致溶酶体肿胀、细胞破坏及脏器功能损害,并引起一系列临床表现。
主要诊断方法
酶学检测:明确外周血白细胞、皮肤成纤维细胞或肌肉组织α-葡萄糖苷酶活性缺乏是诊断GSD II的“金标准”,残留的酶活性水平与临床表现严重程度呈负相关。
GAA基因突变分析:有助于明确诊断,给予有效的遗传咨询和产前诊断。
辅助诊断:血清激酶测定、心脏检查、肺功能测定、肌活检、肌肉影像学检查等。
GSD II治疗预后
GSDⅡ是一个多系统受累的疾病,早期诊断和早期治疗是改善预后的关键。
酶替代治疗:婴儿型及晚发型患者均可使用rhGAA(如Myozyme)。对于婴儿型患者,一旦确诊,应尽早开始酶替代治疗,可显著延长生存期。
对症支持治疗:多学科综合治疗,如心血管系统、呼吸系统、消化系统、运动康复等。
 冀公网安备13109802000548
冀公网安备13109802000548